生信软件 | FastQC(质量控制,查看测序质量)
生信软件 | FastQC
介绍
- 高通量测序数据的高级质控工具
- 输入FastQ,SAM,BAM文件,输出对测序数据评估的网页报告
安装
conda install fastqc
这里需要安装Conda (这是一款用于安装多数生物信息分析软件的管理软件,重要的是可以解决软件依赖问题) : Conda 安装使用图文详解
使用
fastqc -t 12 -o out_path sample1_1.fq sample1_2.fq
-o –outdir:输出路径
–extract:结果文件解压缩
–noextract:结果文件压缩
-f –format:输入文件格式.支持bam,sam,fastq文件格式
-t –threads:线程数
-c –contaminants:制定污染序列。文件格式 name[tab]sequence
-a –adapters:指定接头序列。文件格式name[tab]sequence
-k –kmers:指定kmers长度(2-10bp,默认7bp)
-q –quiet: 安静模式
文档:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
结果解读

完全正常(绿),略有异常(橙) )或异常(红)
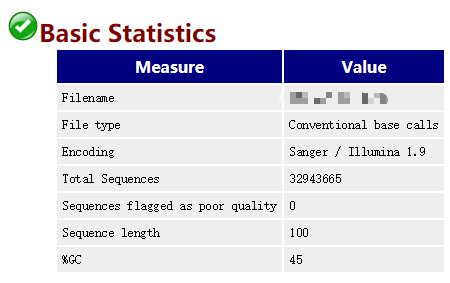
Basic Statistics (基础统计)
Per base sequence quality
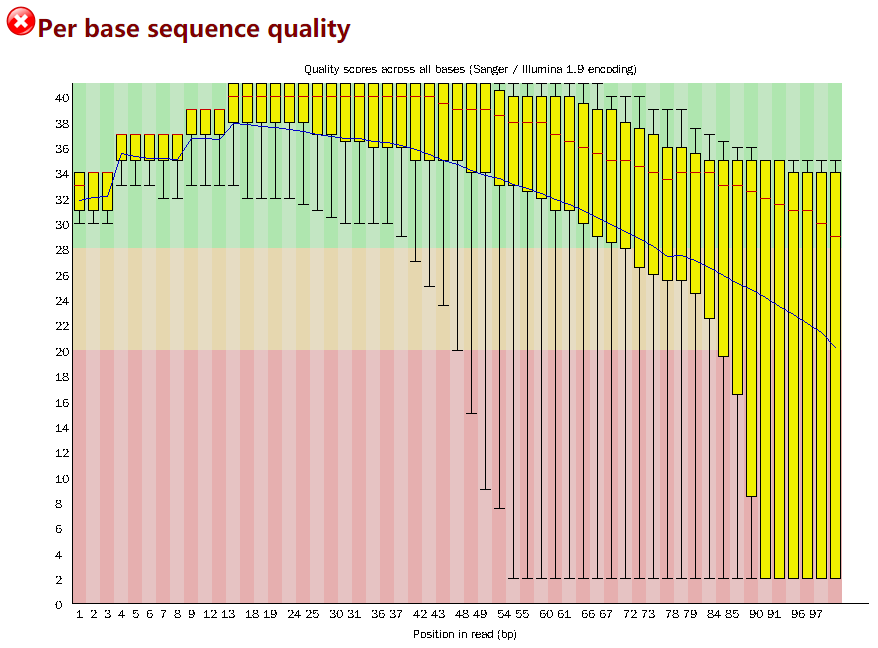
- 这是 read length = 100 的scRNAseq数据,横轴为read位置,纵轴是quality。
- quality = -10*log10(p),p为测错的概率。
- 根据quality给出质量结果:正常区间(28 - 40),警告区间(20-28),错误区间(0-20)。
- 比如,当read的某一位置的p=0.01,quality=20,那么它就处于错误区间。
Per tile sequence quality
Per base quality scores
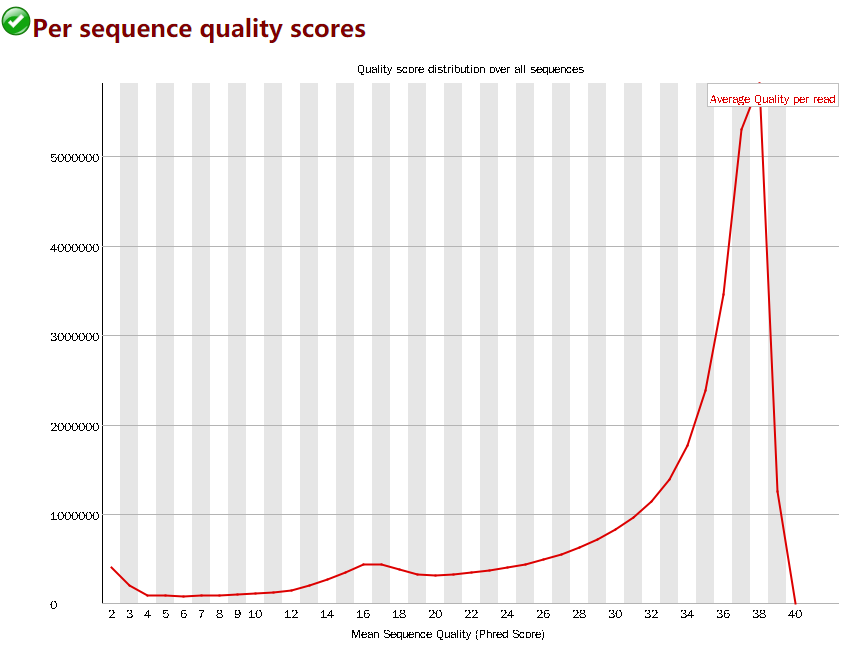
- 横轴为quality,纵轴为reads计数。
- 当峰值处于quality为0-20时,报错。
Per sequence sequence content
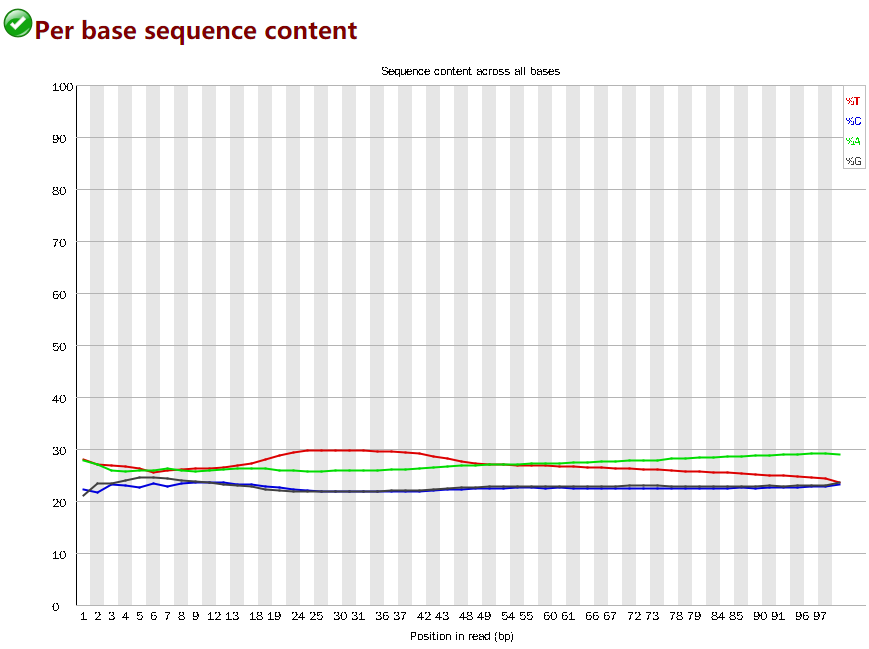
- 横轴为位置,纵轴为百分比
- 正常测序数据为频率相近的四种碱基,无位置差异。表现在图上的话,四条线应该是平行且接近。
- 当任意位置A/T与G/C相差大于10%报警告,大于20%报错
Per base GC content
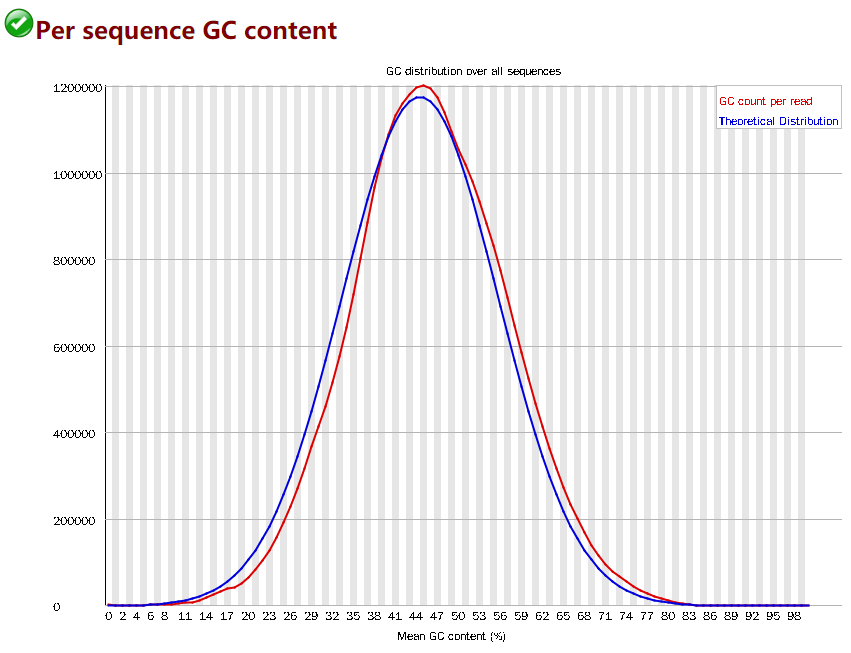
- 横轴为GC含量,纵轴为read计数。红色为实际测得,蓝色为理论分布。
- 如果曲线形状不符,代表文库污染
- 偏离大于15%,报警告;大于30%,报错
Per base N content
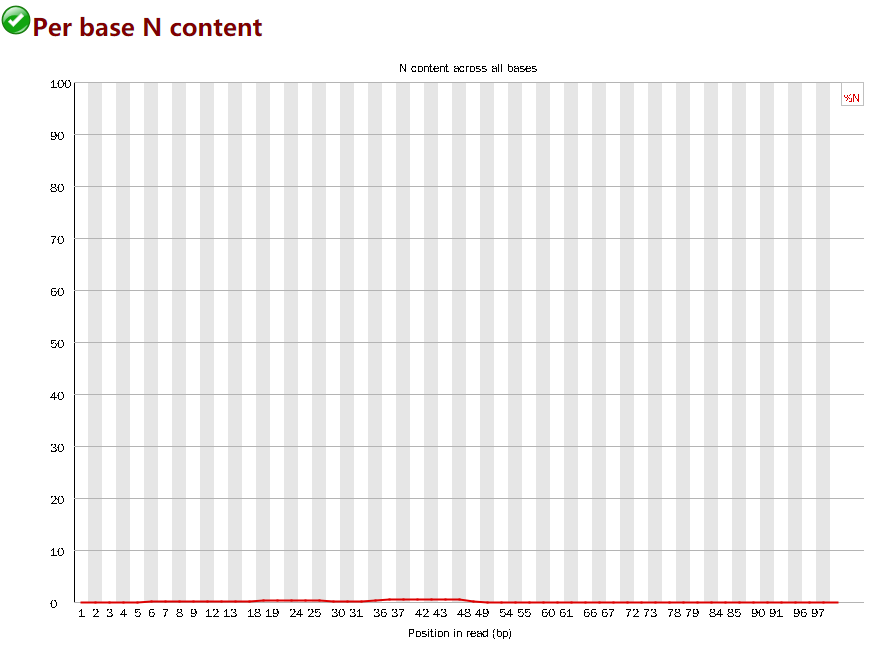
- N 代表测序仪不能识别的碱基,横轴代表read位置,纵轴代表占比
- 如果正常测序,红线应该是趋近与0的直线
- 当任意位置N占比大于5%,报警告;大于20%,报错
Sequence Length Distribution
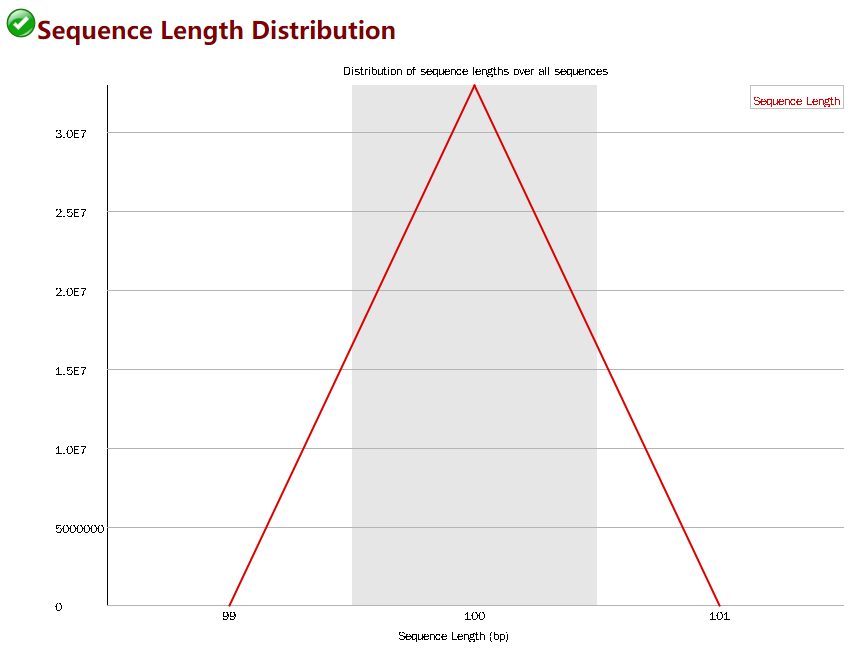
- reads 长度不一致报警告;reads长度为0是报错
Sequence Duplication Levels
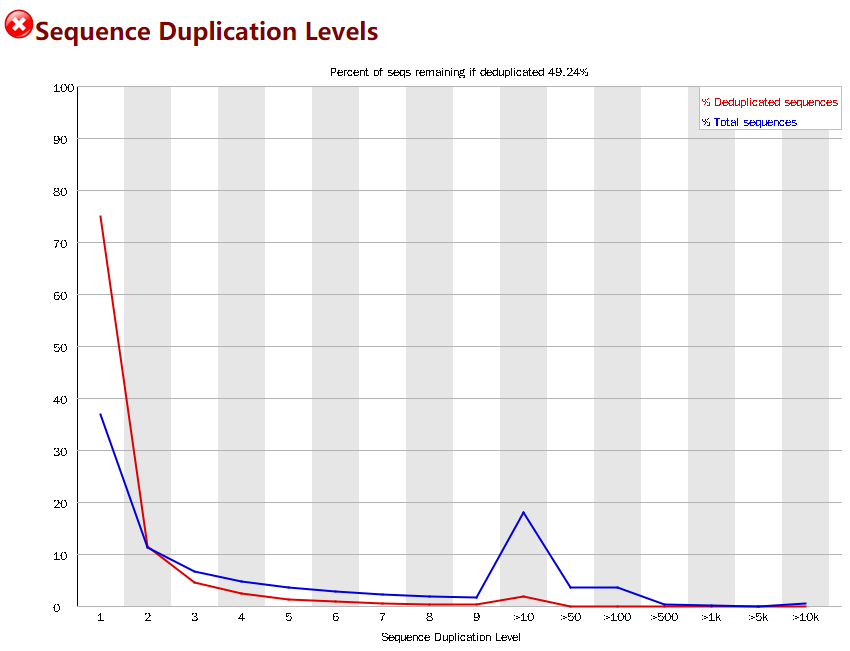
- 横坐标为重复(duplication)的次数,纵坐标为reads的数目,以unique reads的总数作为100%
- 比如,当unique reads数大约为10%时,有两个重复;正常测序开始较高,后续趋近0%
Adapter Content
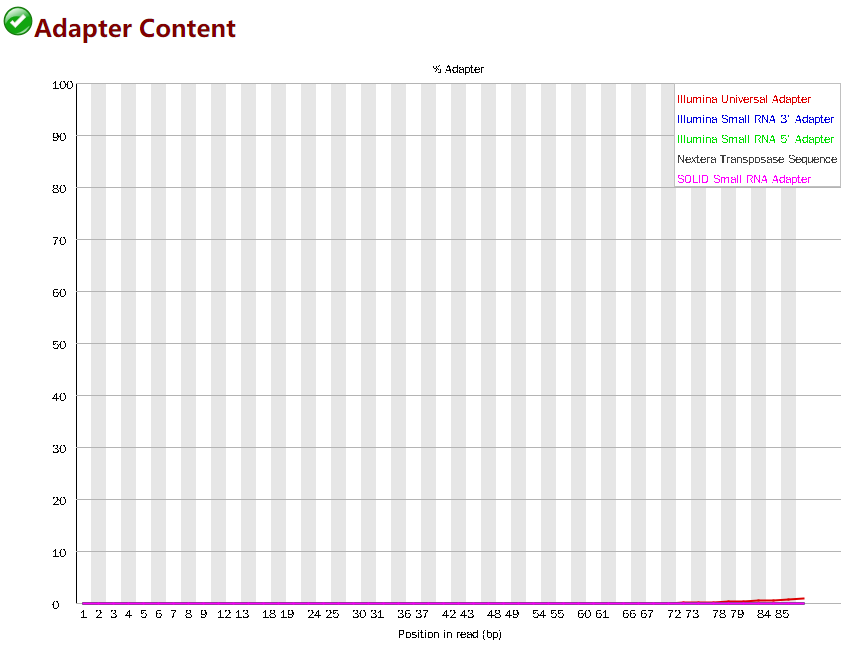
- 横坐标为read位置,纵坐标为Adapter序列占比;如果fastqc默认参数会将所有的常见的Adapter都列出
- 正常情况是趋于0的直线,也就是说序列两端Adapter已经去除干净;如果有Adapter,需要先用cutadapt去接头
本博客所有文章除特别声明外,均采用 CC BY-NC-SA 4.0 许可协议。转载请注明来自 白墨!
 微信
微信 支付宝
支付宝
相关推荐





评论

