生物信息中的Python 01 | 从零开始处理基因序列
一、 序列数据的下载
在开始了解序列的处理流程时,我们先要知道序列下载网址。其中一个知名的网站就是NCBI (National Center for Biotechnology Information)美国国立生物技术信息中心。
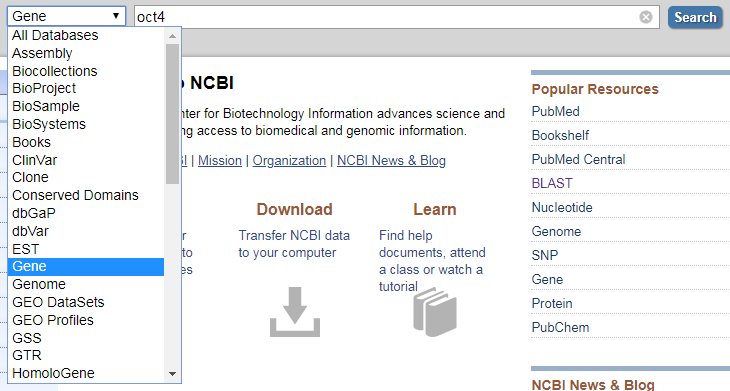
1、通过如下的网站进入 NCBI ,可以看到它包含许多的子库,其中 Gene 就是我们一般下载基因序列的库,接下来,在后面的输入框输入 oct4 并点击 Search。
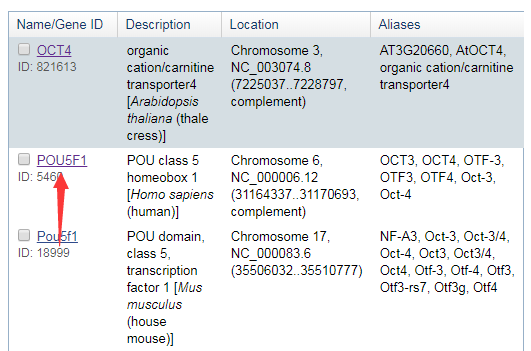
2、可以看到该基因在不同物种和实验中所测得的相同基因序列,我们选择其中智人的POU5F1基因。
值得注意的是 POU5F1 是 Oct4 基因的别名,本质上指的一个基因
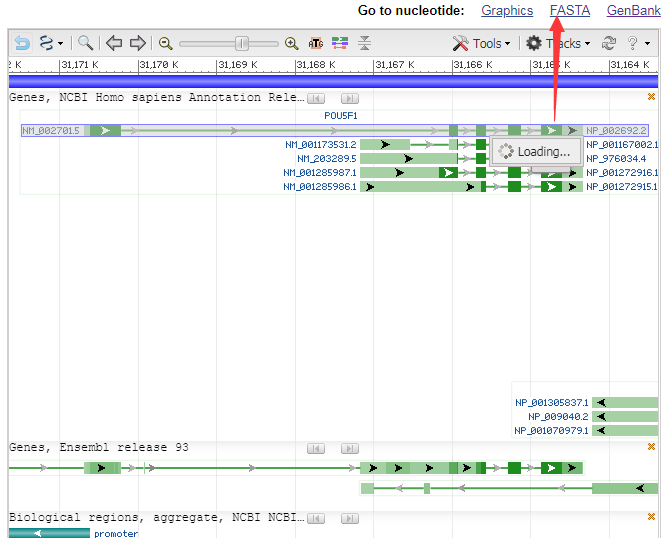
3、向下滚动,直到看到如下图所示的 FASTA 链接,点击进入。
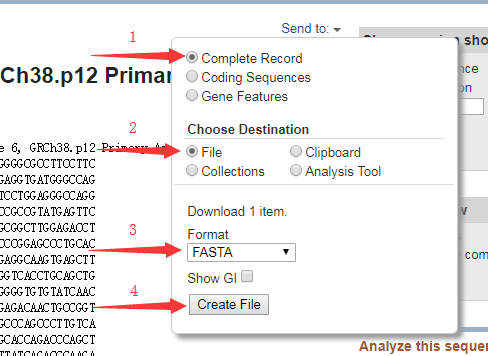
4、在这个页面就可以看到通过测序技术所得到的DNA序列。

5、通过如下步骤我们可以得到该基因序列的 fasta 格式文件
6、你也可以按照上述步骤尝试获取[ Mus musculus ] 的 fasta 序列,我们后面的分析需要用到
二、 DNA序列基本处理
Python版本:Python 3.6
IDE:Pycharm (https://www.jetbrains.com/pycharm/) 下载 Pycharm 的免费社区版就足够我们学习使用
操作系统:Win7
0、在Pycharm里新建如下目录的项目
1、进入main.py文件,我们先把序列文件读取出来看看,到底是怎样的结果
with open('res/sequence1.fasta') as file: |
2、可以看到Fasta格式开始于一个标识符>,然后是一行描述,下面是序列,直到下一个>,表示下一条序列
这些字符串看起来和下载 Fasta 文件页面显示的差不多,但是这不是我们想要的结果

3、接下来我们把描述字段和序列分别提取并存储在字典中
fasta = {} |
用函数把上面的代码装起来,方便后续调用
def get_fasta(fasta_path): |
4、拿到规范化的数据,我们现在来看看具有它具有的生物学意义,这里为了以后方便调用,使用函数的形式来实现
4.1 核苷酸计数,碱基偏好性:
这里的统计数值可以查看碱基偏好性。比如, 一定类型的小RNA会有特定的碱基偏好性,它的第一个碱基偏好U。可以用于评价数据质量。如果miRNA 第一碱基不是U偏好,说明数据或分析过程有问题。
# 核苷酸计数 |
4.2 GC含量:
(A+T)/(G+C)之比随DNA的种类不同而异。GC含量愈高,DNA的密度也愈高,同时热及碱不易使之变性,因此利用这一特性便可进行DNA的分离或测定。同时,物种的GC含量有着特异性,以此可以判断测序后的数据是否合格。
# CG 含量 |
4.3 DNA 翻译为 RNA:
# DNA 翻译为 RNA |
4.4 RNA 翻译为 蛋白质:
def rna_trans_protein(rnaSeq): |
4.5 获取反向序列:
# 获取反向序列 |
4.6 最后我们来一个main来把上面的函数统统运行一遍
if __name__ == '__main__': |
部分结果如下:
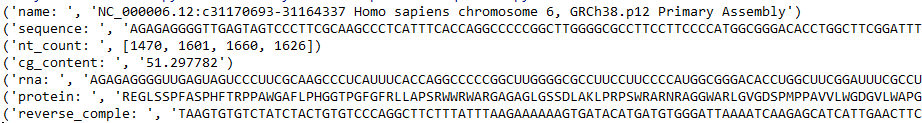
了解基因结构
生物信息中的Python 02 | 用biopython解析序列
生物信息中的Python 03 | 自动化操作NCBI
生物信息中的Python 04 | 批量下载基因与文献
本博客所有文章除特别声明外,均采用 CC BY-NC-SA 4.0 许可协议。转载请注明来自 白墨!
 微信
微信 支付宝
支付宝
相关推荐






评论

