UCSC 基因组浏览器配置详解
一、配置参数
UCSC基因组浏览器:传送门
1、点击配置
2、进入配置页面:
点击刚刚运行的文件 BedGraph Format
2、轨迹配置页面
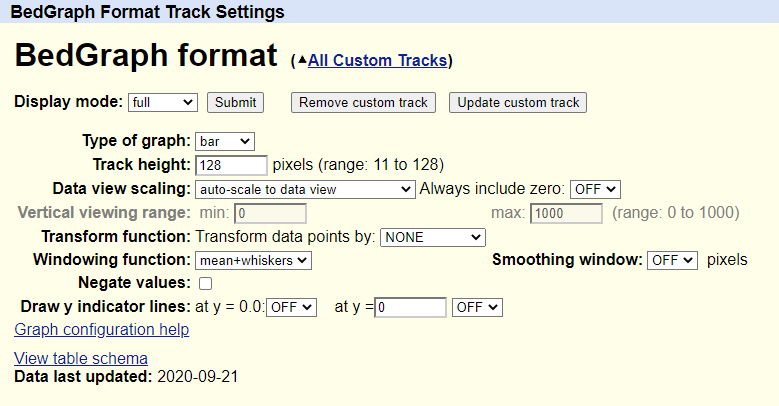
- Type of graph :默认以
bar,条形图来显示,选择point会以点或线来显示 - Track height :设置图形高度,像素为单位
- Data view scaling (boxed in red) :
- 如果选中
use vertical viewing range setting选项,将使用Vertical viewing range设置中指定的参数显示数据 - 如果选中
auto-scale to data view选项,将图形配置为自动缩放到当前视图中最小和最大数据点定义的范围。要在选择自动缩放时,始终保持 y = 0 ,需要Always include zero设置为ON。
- 如果选中
查看复合组中的信号轨迹时,请使用group auto-scale 功能,以使所有轨迹相对于当前视图中具有最大最大数据点的组中的一个轨迹进行缩放。
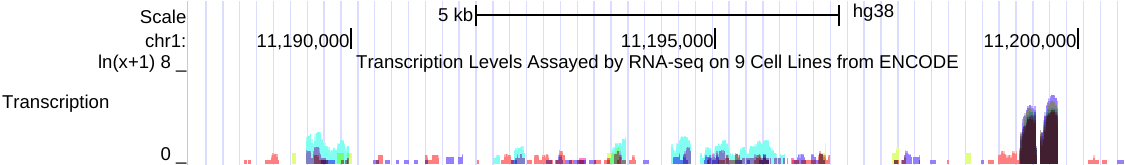
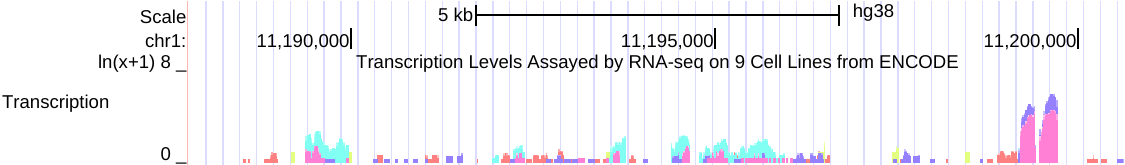
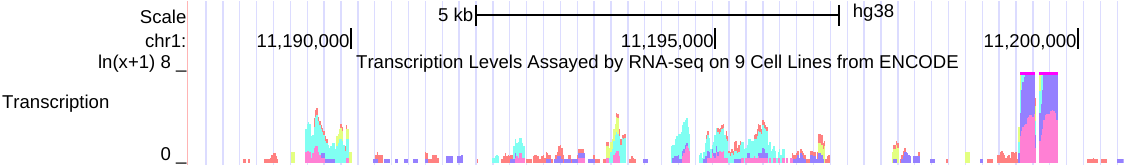
例如,以下是在相关RNA-seq实验的组合中,来自多个细胞系的同一数据的两个视图的并排图像。
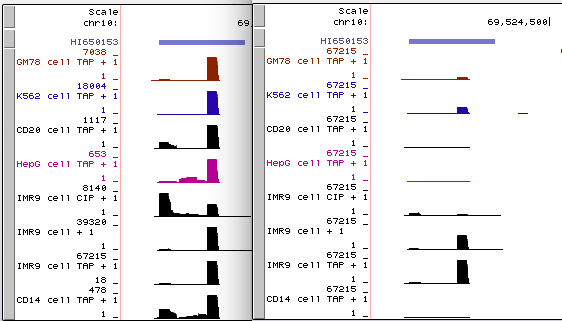
左侧(点击查看)是原始的 auto-scale to data view 设置,其中每个轨迹都自动缩放到该轨迹的最高值。
右侧(点击查看)是针对相同RNA-seq数据的 group auto-scale 设置,其中所有轨迹相对于具有最高值(IMR9细胞TAP +1的67215)的一个轨迹进行缩放。
Transform function :通过下拉菜单中选择的功能转换数据点。通常,默认设置为“无”
Windowing function :当视图太大而无法显示单个数据值时,必须将这些值组合起来以产生一个绘图点。此选项指定要使用的合并功能(默认为“均值”):
- Mean+whiskers:在深色阴影下显示平均值,在中等阴影下显示均值周围的一个标准偏差,在浅色阴影下显示最大值/最小值。对于条形图,只有平均值,平均值加上标准偏差和最大值可见。如果是叠加方法,则此模式不可用。
- Maximum:显示所有要合并的点的最大值
- Mean:显示平均值
- Minimum: 显示所有要组合点的最小
Smoothing window :等效于图形上的趋势线计算。默认设置为“关”。 设置数值用于确定要在图形上传递以平滑条或线边缘的平滑窗口的大小,以像素为单位。
Negate values:取反,选中后,所有值都取反,这意味着正值变为负值,反之亦然。这对于表示负链上的转录等非常有用。
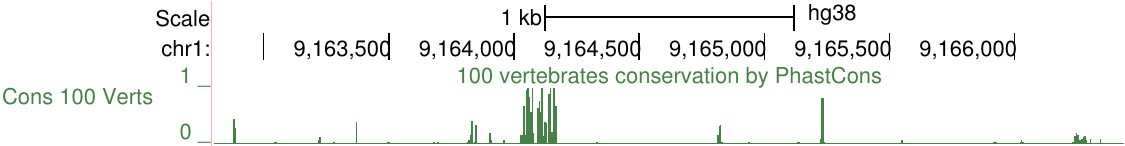
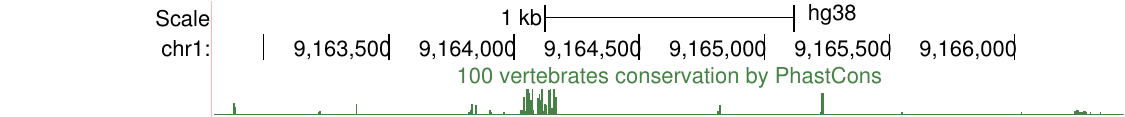
比如,下图显示了不同链上两个基因SIRT1和HERC4周围的ENCODE RNA-seq数据,负信号轨迹使用取反值,显示以强调HERC4在负链上表达。该图像还显示以点为单位绘制的信号和16像素的平滑窗口。

Draw y indicator lines :
当** y = 0.0 **时:选择
ON以显示在图形上标记 0.0 位置的线(默认为 OFF)当y= :选择
ON设置以指定的数值在图形上显示一条线(默认值为0和OFF)。这条线可以用来标记图形上的重要阈值。例如,在下面的图像中, y = 3。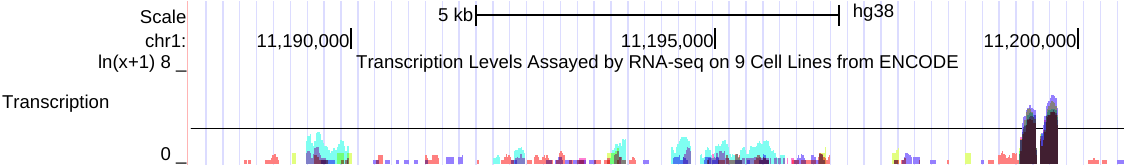
二、轨迹显示
1、显示模式
Dense
显示的轨迹将所有特征折叠为一行。线条颜色越深,该位置的摆动值越大

Squish
轨迹显示时所有特征都折叠成一行,非常类似于具有更大压缩率的 Dense 显示模式

Full
轨迹显示与每个注释功能关联的 wiggle 值,从而创建类似直方图的图像
Pack
轨迹显示与每个注释功能关联的 wiggle 值,从而创建类似于直方图的图像,这与具有更大压缩率的完整 Full 模式非常相似
Hide
不显示轨迹
2、叠加方法(Overlay)
并非所有基于图形的轨迹都包括 Overlay 选项
Transparent
此设置显示多个子轨迹的彩色透明图形,并叠加在同一垂直空间中
Solid
此设置显示多个子轨迹的彩色不透明图形,然后叠加在同一垂直空间中
Stacked
此设置显示每个堆叠在一起的图形,其中图形的最高点是所有值的总和
None
此设置将每个图形显示在其自己的独立的垂直空间中

三、常用文件
bigwig 文件绘制轨道
1、加入自定义轨道:https://genome.ucsc.edu/cgi-bin/hgCustom
|
2、绘制出轨道
wig 文件绘制轨道
1、下载数据:
wiggle 文件:http://genome.ucsc.edu/goldenPath/help/examples/wigVarStepExample.gz
chrom.sizes 文件:http://genome.ucsc.edu/goldenPath/help/hg19.chrom.sizes
2、运行命令:
wigToBigWig wigVarStepExample.gz hg19.chrom.sizes myBigWig.bw
结果会生成 myBigWig.bw 文件
3、将生成的 bigWig 文件放在可web访问的服务器:
http://bioinfo.ziptop.top/myBigWig.bw
4、绘制出轨道
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-SkPuw5fh-1603975335831)(http://baimoc.ziptop.top/blog/20200921/IEtD1mvTJk32.png)]
bedGraph 文件绘制轨道
1、新建bedGraph 文件,
必须为每个数据轨道创建一个单独的 bedGraph 文件,比如in.bedGraph:
|
2、将 bedGraph 转换为 BigWig 文件:
bedGraphToBigWig 下载地址:http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/bedGraphToBigWig
bedGraphToBigWig in.bedGraph chrom.sizes bgBigWig.bw
bedGraphToBigWig程序不接受压缩的bedGraph输入文件
3、将生成的 bigWig 文件放在可web访问的服务器:
http://bioinfo.ziptop.top/bgBigWig.bw
4、输入轨道地址,提交
http://genome.ucsc.edu/cgi-bin/hgCustom
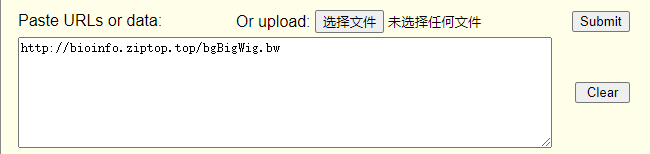
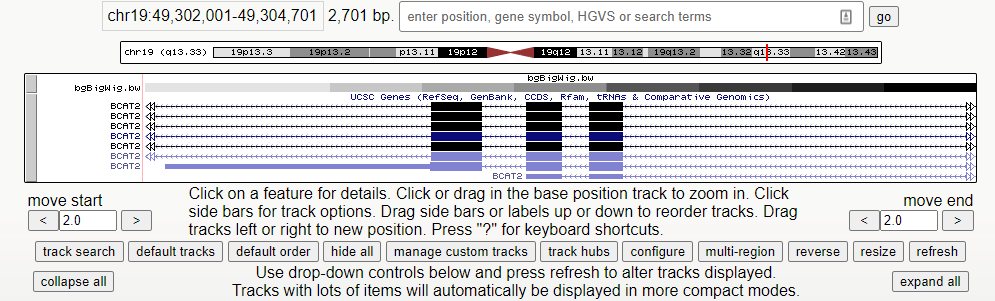
6、绘制出轨道
 微信
微信 支付宝
支付宝







