生信格式 | bigwig,bw (基因组浏览器绘制)
一、特点及适用场景:
- 存放区间的坐标轴信息(如染色质可及性,转录因子结合区域)和相关评分(score)的文件,主要用于存储密集,连续的数据
- 主要用于在基因组浏览器上查看数据的连续密度图
- wig或bedGraph的索引二进制文件,也就是可以由这两种文件转换得到
- 后缀名:
.bw,.bigwig - 在处理大型数据集时,bigWig文件的显示性能比常规的wig文件快得多
- 数据必须是连续的并且由大小相等的元素组成,如果数据是稀疏或包含大小不同的元素时,请使用bedGraph格式
二、wig 转 bigwig
BigWig文件可以使用wigToBigWig程序从wiggle(wig)格式文件转换得到
1、 创建 wig 文件
wig 文件转换为bigWig文件时,必须为每个数据轨迹创建一个单独的 wig 文件。
从 wig 文件中删除任何现有的“ track”或“ browser”行,使其仅包含数据。
文件名命名为 input.wig
|
2、创建chrom.sizes文件
wget http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/fetchChromSizes
fetchChromSizes hg38 > hg38.chrom.sizes
3、转换为 bigWig 文件
http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/wigToBigWig
wigToBigWig input.wig hg38.chrom.sizes BigWig.bw
4、将生成的 bigwig 文件放在web可访问的地址
这里提供了两种方式:
- Track Hub 是官方提供 Web 可访问的基因组数据目录
- 自己搭建个网站,把数据开放给外部,比如,我生成的bigwig文件链接放在这个url:http://bioinfo.ziptop.top/BigWig.bw
bigWig文件保留在本地可通过Web访问的服务器(http,https或ftp)上,并且仅将当前显示的染色体位置所需的部分,才会成为本地缓存的“稀疏文件”。
如果无权访问Web服务器并且需要bigWig文件的托管空间,请参阅Track Hub帮助文档的“ 托管”部分。
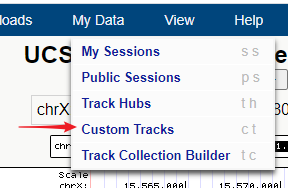
5、 选择菜单栏My Data 的Custom Tracks
6、将上面的代码粘贴到输入框,点 Submit
http://bioinfo.ziptop.top/BigWig.bw

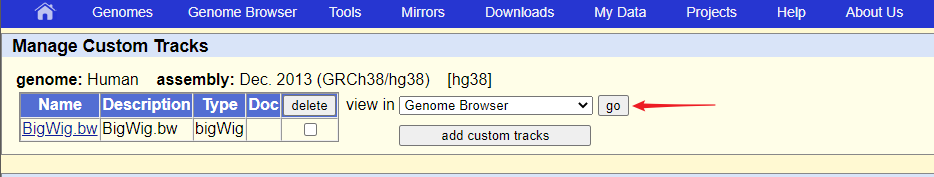
7、点 go
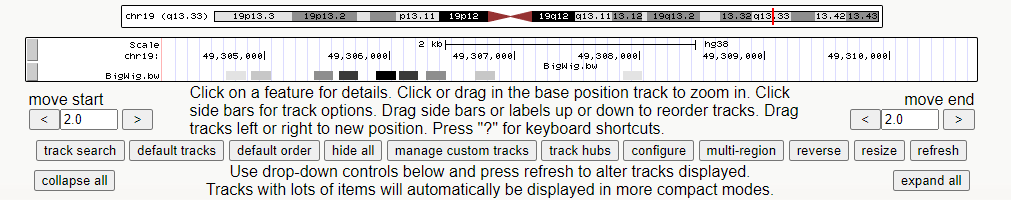
8、在基因组浏览器中绘制的轨迹
可以看到参考基因组相应的位置,不同的值用不同颜色代表
9、定制轨迹线参数
默认情况下,将使用文件名来命名轨迹。要配置轨迹标签或其他可视化选项,必须创建一条轨迹
|
粘贴上面的代码,点 Submit
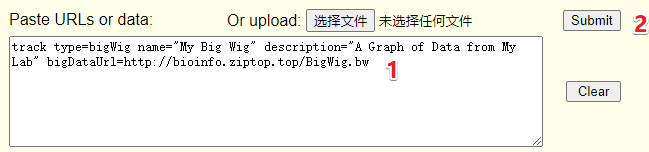
10、轨迹列表,点 go
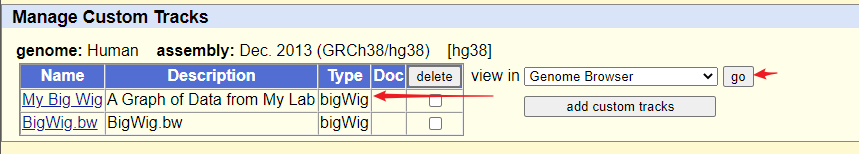
其他参数
|
三、bedGraph 转 bigwig
bedGraphToBigWig程序比未压缩的bedGraph输入文件使用的RAM多大约25% 。
类似于上述过程,这里简写
wget http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/fetchChromSizes
fetchChromSizes hg38 > hg38.chrom.sizes
wget http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/bedGraphToBigWig
bedGraphToBigWig in.bedGraph hg38.chrom.sizes out.bw
如果报错染色体长度超出,需要先剪切bed
用到的工具:wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedClipbedClip input.bed hg38.chrom.sizes output.bed
四、其他工具
http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
bigWigToBedGraph:将bigWig文件转换为 bedGraph 格式。bigWigToWig:将bigWig文件转换为 wig 格式。注意:如果从bedGraph创建了bigWig文件,则bigWigToWig会将文件还原回bedGraph。bigWigSummary:从bigWig文件中提取摘要信息。bigWigAverageOverBed:计算每个 bed 上可能有内含子的bigWig的平均得分。bigWigInfo:打印出有关bigWig文件的信息。
 微信
微信 支付宝
支付宝







