一、安装 如果没有conda 基础,参考: Conda 安装使用图文详解(2021版)
pip install scanpy
二、使用 1、准备工作 import numpy as npimport pandas as pdimport scanpy as sc3 80 , facecolor='white' )'write/pbmc3k.h5ad' './filtered_gene_bc_matrices/hg19/' , 'gene_symbols' , True )
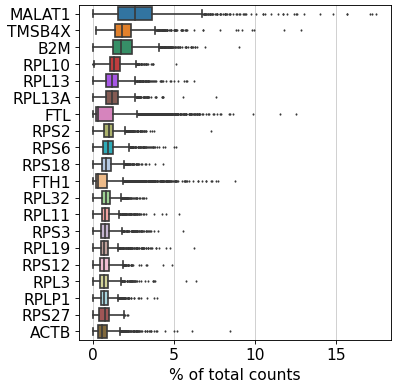
2、预处理 显示在所有细胞中在每个单细胞中产生最高计数分数的基因
sc.pl.highest_expr_genes(adata, n_top=sc.pl.highest_expr_genes(adata, n_top=20 , )
过滤低质量细胞样本 过滤在少于三个细胞中表达,或一个细胞中表达少于200个基因的细胞样本
sc.pp.filter_cells(adata, min_genes=200 )sc.pp.filter_cells(adata, min_genes=200 )3 )
过滤包含线粒体基因和表达基因过多的细胞
线粒体基因的转录本比单个转录物分子大,并且不太可能通过细胞膜逃逸。因此,检测出高比例的线粒体基因,表明细胞质量差(Islam et al. 2014; Ilicic et al. 2016)。
表达基因过多可能是由于一个油滴包裹多个细胞,从而检测出比正常检测要多的基因数,因此要过滤这些细胞。
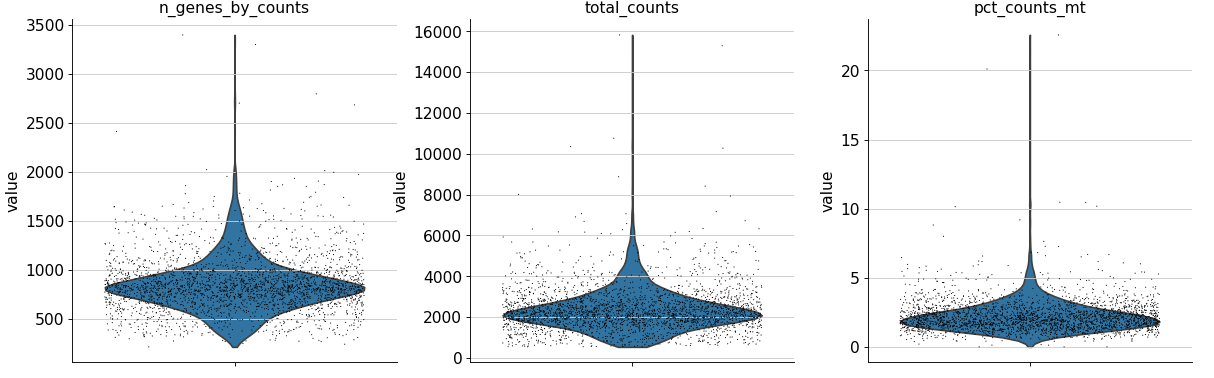
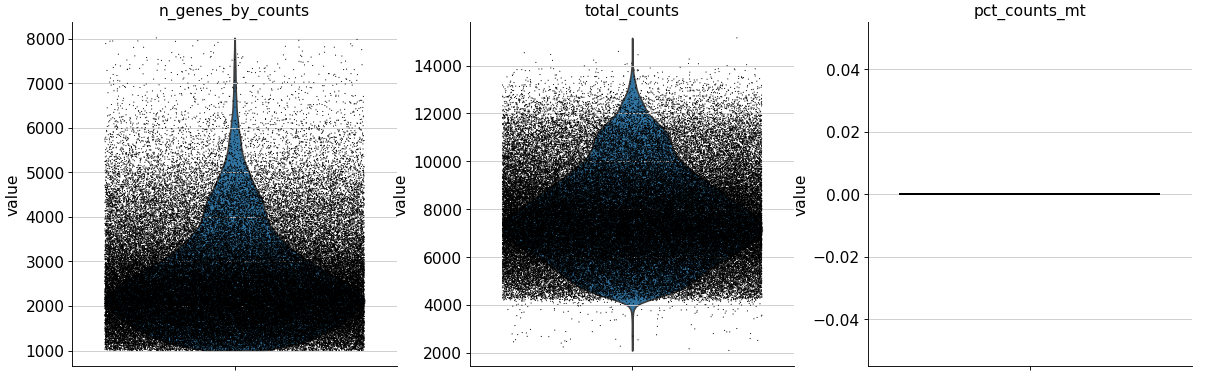
adata.var['mt' ] = adata.var_names.str .startswith('MT-' ) 'mt' ], percent_top=None , log1p=False , inplace=True )'n_genes_by_counts' , 'total_counts' , 'pct_counts_mt' ],0.4 , multi_panel=adata.var['mt' ] = adata.var_names.str .startswith('MT-' ) 'mt' ], percent_top=None , log1p=False , inplace=True )'n_genes_by_counts' , 'total_counts' , 'pct_counts_mt' ],0.4 , multi_panel=True )
生成的三张小提琴图代表:表达基因的数量,每个细胞包含的表达量,线粒体基因表达量的百分比。
过滤
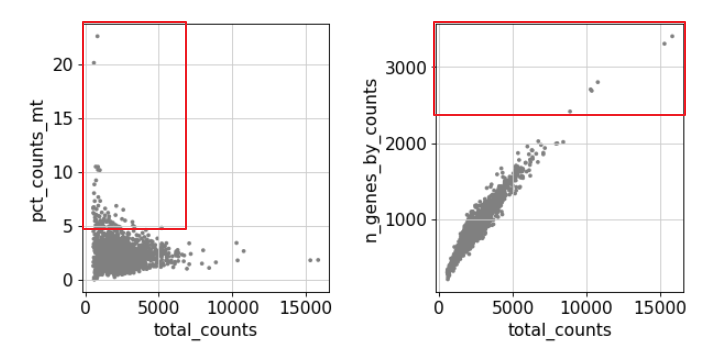
sc.pl.scatter(adata, x='total_counts' , y='pct_counts_mt' )'total_counts' , y=sc.pl.scatter(adata, x='total_counts' , y='pct_counts_mt' )'total_counts' , y='n_genes_by_counts' )
过滤线粒体基因表达过多或总数过多的细胞,也就是红框标识的样本。
5 , :]5 , :]2500 , :]
3、检测特异性基因 归一化
1e4 )
存储数据
将 AnnData 对象的 .raw 属性设置为归一化和对数化的原始基因表达,以便以后用于基因表达的差异测试和可视化。这只是冻结了 AnnData 对象的状态。
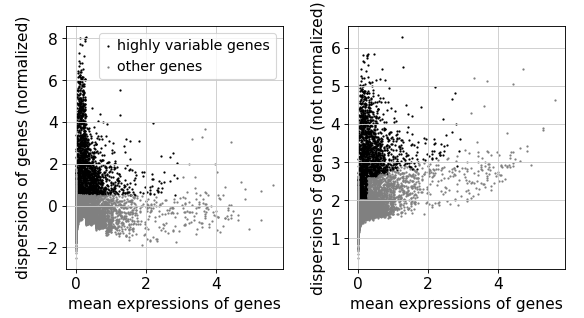
识别特异性基因
# 计算
获取只有特异性基因的数据集
'total_counts' , 'pct_counts_mt' ])'total_counts' , 'pct_counts_mt' ])10 )
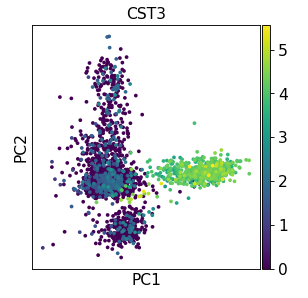
4、主成分分析(Principal component analysis) 通过运行主成分分析 (PCA) 来降低数据的维数,可以对数据进行去噪并揭示不同分群的主因素。
'arpack' )'arpack' )'CST3' )
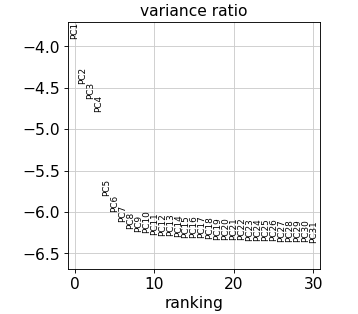
检查单个 PC 对数据总方差的贡献,这可以提供给我们应该考虑多少个 PC 以计算细胞的邻域关系的信息,例如用于后续的聚类函数 sc.tl.louvain() 或 tSNE 聚类 sc.tl.tsne()。
sc.pl.pca_variance_ratio(adata, log=sc.pl.pca_variance_ratio(adata, log=True )
5、领域图,聚类图(Neighborhood graph) 使用数据矩阵的 PCA 表示来计算细胞的邻域图。为了重现 Seurat 的结果,我们采用以下值。
建议使用 UMAP ,它可能比 tSNE 更忠实于流形的全局连通性,因此能更好地保留轨迹。
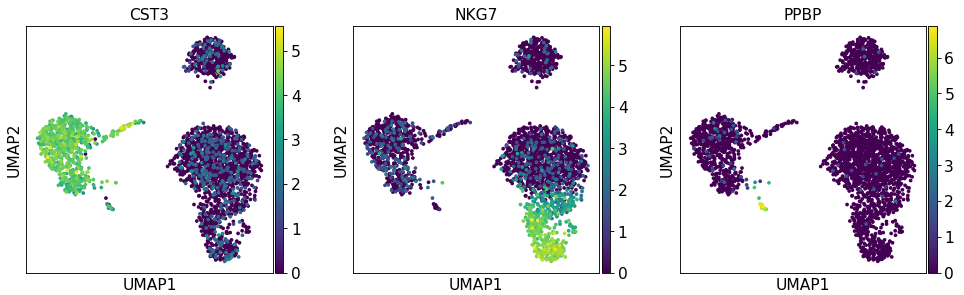
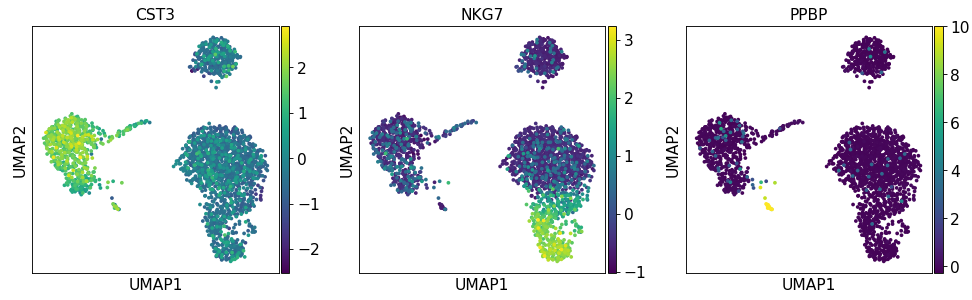
sc.pp.neighbors(adata, n_neighbors=10 , n_pcs=40 )'CST3' , 'NKG7' , sc.pp.neighbors(adata, n_neighbors=10 , n_pcs=40 )'CST3' , 'NKG7' , 'PPBP' ])
为了绘制缩放矫正的基因表达聚类图,需要使用 use_raw=False 参数。
sc.pl.umap(adata, color=['CST3' , 'NKG7' , 'PPBP' ], use_raw=sc.pl.umap(adata, color=['CST3' , 'NKG7' , 'PPBP' ], use_raw=False )
目前还没有计算出各个细胞类群,下面进行聚类
Leiden 图聚类
'leiden' ])
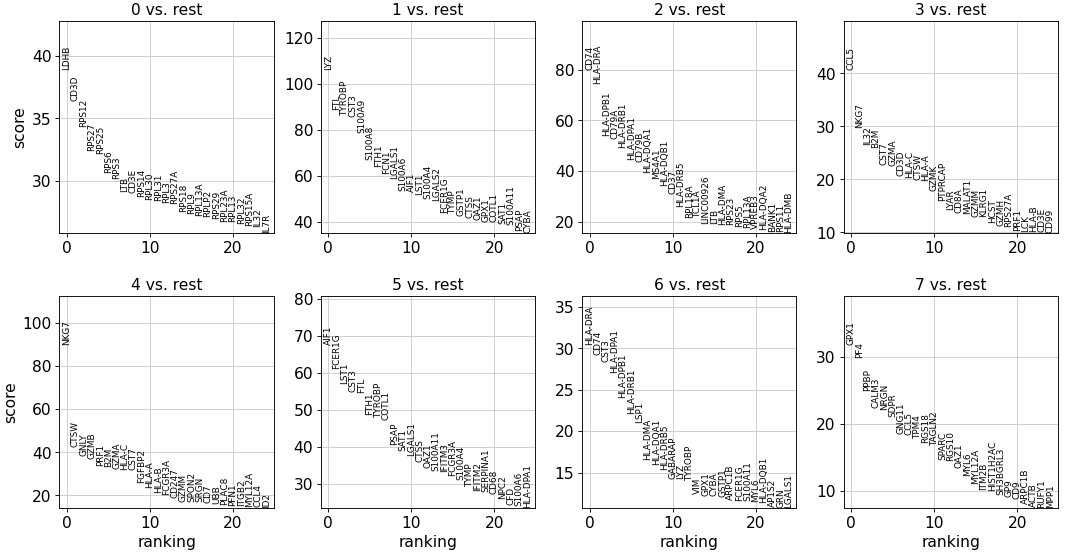
6、检索标记基因 先计算每个 leiden 分群中高度差异基因的排名,取排名前 25 的基因。
默认情况下,使用 AnnData 的 .raw 属性。
T-test
最简单和最快的方法是 t 检验。
sc.tl.rank_genes_groups(adata, 'leiden' , method='t-test' )25 , sharey=sc.tl.rank_genes_groups(adata, 'leiden' , method='t-test' )25 , sharey=False )
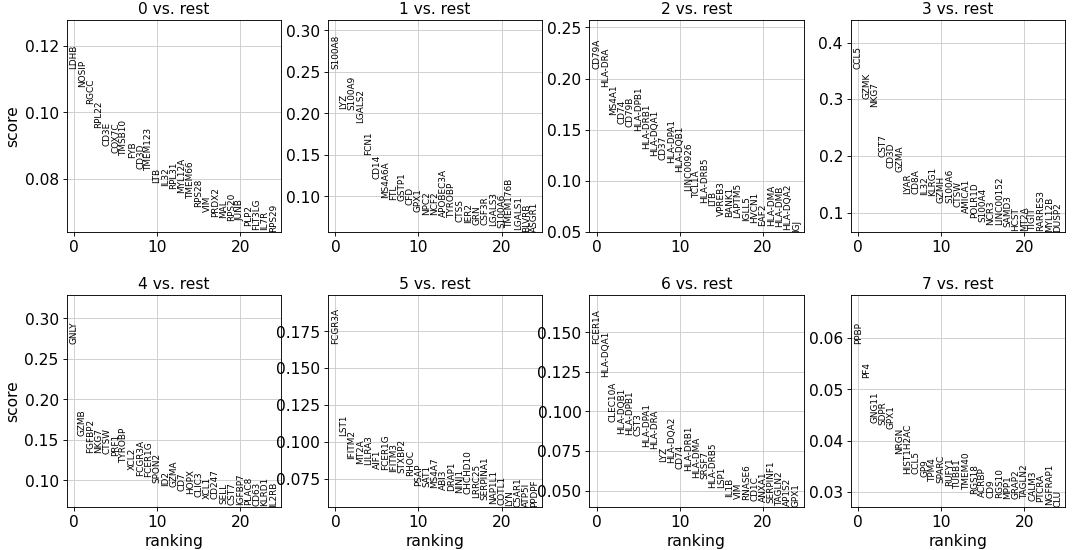
Wilcoxon rank-sum
Wilcoxon rank-sum (Mann-Whitney-U) 检验的结果非常相似,还可以使用其他的差异分析包,如 MAST、limma、DESeq2 和 diffxpy。
sc.tl.rank_genes_groups(adata, 'leiden' , method='wilcoxon' )25 , sharey=False )
逻辑回归
sc.tl.rank_genes_groups(adata, 'leiden' , method='logreg' )25 , sharey=sc.tl.rank_genes_groups(adata, 'leiden' , method='logreg' )25 , sharey=False )
使用逻辑回归对基因进行排名 Natranos et al. (2018),这里使用多变量方法,而传统的差异测试是单变量 Clark et al. (2014)
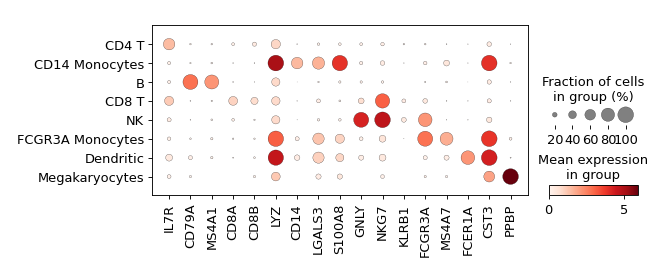
除了仅由 t 检验发现的 IL7R 和由其他两种方法发现的 FCER1A 之外,所有标记基因都在所有方法中都得到了重现。
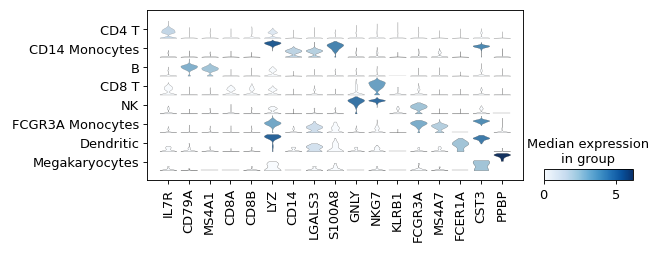
Louvain Group
Markers
Cell Type
0
IL7R
CD4 T cells
1
CD14, LYZ
CD14+ Monocytes
2
MS4A1
B cells
3
CD8A
CD8 T cells
4
GNLY, NKG7
NK cells
5
FCGR3A, MS4A7
FCGR3A+ Monocytes
6
FCER1A, CST3
Dendritic Cells
7
PPBP
Megakaryocytes
根据已知的标记基因,定义一个标记基因列表供以后参考:
marker_genes = ['IL7R' , 'CD79A' , 'MS4A1' , 'CD8A' , 'CD8B' , 'LYZ' , 'CD14' ,'LGALS3' , 'S100A8' , 'GNLY' , 'NKG7' , 'KLRB1' ,'FCGR3A' , 'MS4A7' , 'FCER1A' , 'CST3' , marker_genes = ['IL7R' , 'CD79A' , 'MS4A1' , 'CD8A' , 'CD8B' , 'LYZ' , 'CD14' ,'LGALS3' , 'S100A8' , 'GNLY' , 'NKG7' , 'KLRB1' ,'FCGR3A' , 'MS4A7' , 'FCER1A' , 'CST3' , 'PPBP' ]
载入数据
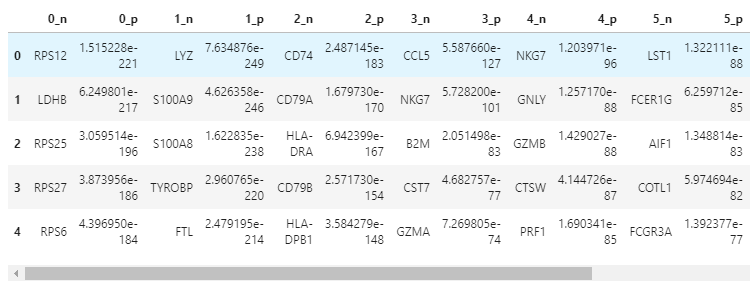
获取聚类分组和分数
result = adata.uns['rank_genes_groups' ]'names' ].dtype.names'_' + key[:1 ]: result[key][group]for group in groups for key in ['names' , 'pvals' ]}).head(result = adata.uns['rank_genes_groups' ]'names' ].dtype.names'_' + key[:1 ]: result[key][group]for group in groups for key in ['names' , 'pvals' ]}).head(5 )
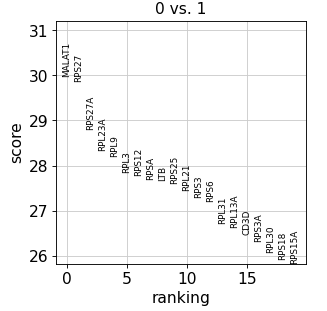
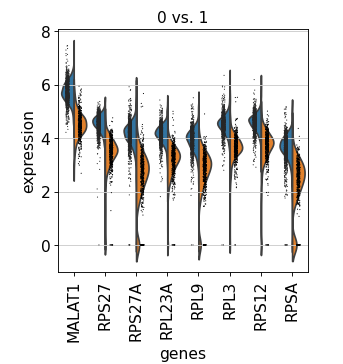
Group 1 与 Group 2 比较,进行差异分析
sc.tl.rank_genes_groups(adata, 'leiden' , groups=['0' ], reference='1' , method='wilcoxon' )'0' ], n_genes=sc.tl.rank_genes_groups(adata, 'leiden' , groups=['0' ], reference='1' , method='wilcoxon' )'0' ], n_genes=20 )
sc.pl.rank_genes_groups_violin(adata, groups='0' , n_genes=sc.pl.rank_genes_groups_violin(adata, groups='0' , n_genes=8 )
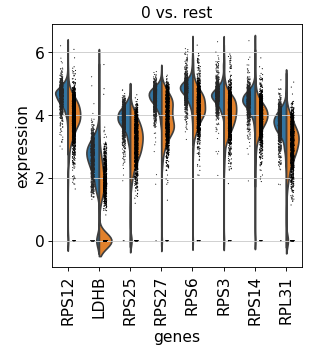
Group 0 与其余组的比较进行差异分析
adata = sc.read(results_file)'0' , n_genes=adata = sc.read(results_file)'0' , n_genes=8 )
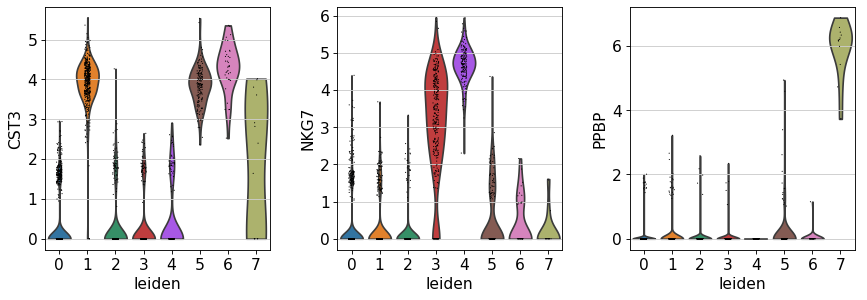
跨类群比较基因
sc.pl.violin(adata, ['CST3' , 'NKG7' , 'PPBP' ], groupby=sc.pl.violin(adata, ['CST3' , 'NKG7' , 'PPBP' ], groupby='leiden' )
根据已知的细胞标记,注释细胞类型
new_cluster_names = ['CD4 T' , 'CD14 Monocytes' ,'B' , 'CD8 T' ,'NK' , 'FCGR3A Monocytes' ,'Dendritic' , 'Megakaryocytes' ]'leiden' , new_cluster_names)'leiden' , legend_loc='on data' , title='' , frameon=False , save=new_cluster_names = ['CD4 T' , 'CD14 Monocytes' ,'B' , 'CD8 T' ,'NK' , 'FCGR3A Monocytes' ,'Dendritic' , 'Megakaryocytes' ]'leiden' , new_cluster_names)'leiden' , legend_loc='on data' , title='' , frameon=False , save='.pdf' )
可视化每个类群的标记基因
气泡图显示:
sc.pl.dotplot(adata, marker_genes, groupby=sc.pl.dotplot(adata, marker_genes, groupby='leiden' );
小提琴图显示
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden' , rotation=sc.pl.stacked_violin(adata, marker_genes, groupby='leiden' , rotation=90 );
7、保存数据 保存压缩文件
如果只想将其用于可视化的人共享此文件,减少文件大小的一种简单方法是删除缩放和校正的数据矩阵。
adata.write(results_file, compression=adata.write(results_file, compression='gzip' )
保存为 h5ad 数据
adata.raw.to_adata().write(adata.raw.to_adata().write('./write/pbmc3k_withoutX.h5ad' )
读取使用 adata = sc.read_h5ad(‘./write/pbmc3k_withoutX.h5ad’)
导出数据子集
'n_counts' , 'louvain_groups' ]].to_csv('./write/pbmc3k_corrected_louvain_groups.csv' )'X_pca1' , 'X_pca2' ]].to_csv('n_counts' , 'louvain_groups' ]].to_csv('./write/pbmc3k_corrected_louvain_groups.csv' )'X_pca1' , 'X_pca2' ]].to_csv('./write/pbmc3k_corrected_X_pca.csv' )
8、番外 我之前在处理较多数据量的时候,会有些地方不一样,具体每个数据集的处理也会有比较大的自由度,比如:
在检测线粒体基因时,这里在质控时,已经把线粒体基因直接剔除。
在做 UMAP 时,可以看到一些类群间的联系和轨迹。
做 TSNE时,可以看到类群间比较干净利索,整体比较“饱满”。
其他 Scanpy 的使用教程:scanpy 单细胞分析包图文详解 01 | 深入理解 AnnData 数据结构


 微信
微信 支付宝
支付宝




