生信软件 | STAR(测序序列与参考序列比对)
零、介绍
- STAR (Spliced Transcripts Alignment to a Reference),用于将测序的 Read 对齐到参考基因组的比对软件,常用于 RNAseq。
- 因其具有较高的准确率,映射速度较其他比对软件高 50 多倍,因此作为 ENCODE 项目的御用 pipeline 工具。
- 它需要占用大量内存,对计算资源有较高的要求。
- STAR 的默认参数针对哺乳动物基因组进行了优化
一、安装
|
二、使用
1、建立索引
|
参数:
- –runMode genomeGenerate:基因组生成模式
- –runThreadN:启用线程数
- –genomeDir:索引输出路径
- –genomeFastaFiles:参考基因组路径
- –sjdbGTFfile:参考基因组注释文件
- –sjdbOverhang:对于不同长度的读取,理想值为
--sjdbOverhangmax(ReadLength)-1。在大多数情况下,默认值 100 与理想值类似。
2、STAR 比对
|
参数:
- –runThreadN:启用线程数
- –genomeDir:索引路径
- –readFilesIn:输入 fastq 的文件路径
- –outSAMtype BAM SortedByCoordinate:输出排序的 bam 文件
- –outFileNamePrefix:输出文件前缀
STAR 的默认参数针对哺乳动物基因组进行了优化。**其他物种可能需要对某些对齐参数进行重大修改,尤其具有较小内含子的生物,必须减小最大和最小内含子大小
三、原理
STAR 的比对算法需要两步:
- 种子搜索
- 聚类,拼接,评分
种子搜索
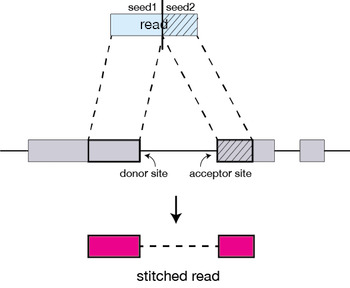
STAR 先搜索与参考基因组上,一个或多个位置完全匹配的最长序列。这些最长的匹配序列称为最大可映射前缀 (Maximal Mappable Prefix,**MMP):
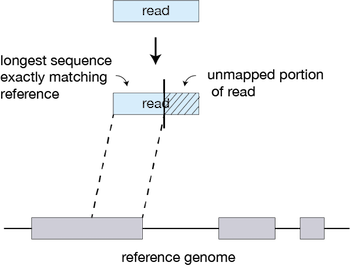
匹配到的 Read 的不同部分称为“seed”。所以对齐到基因组的第一个 MMP 称为seed1。
随后 STAR 将再次仅搜索读数的未映射部分,以找到与参考基因组完全匹配的下一个最长序列 MMP,即seed2,以此类推。
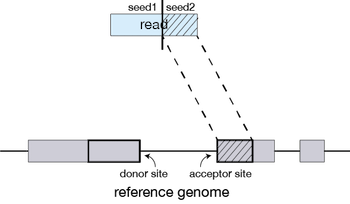
这种 Read 顺序搜索是 STAR 算法效率的基础。
STAR 使用未压缩的后缀数组 (Suffix Array,SA) 来有效搜索 MMP,这允许针对最大的参考基因组进行快速搜索。其他较慢的比对软件使用的算法通常在拆分 Read 和执行比对之前搜索整个 Read 序列。
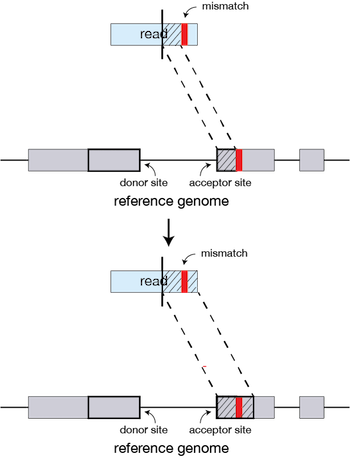
意外情况是:
- 由于不匹配或缺失,STAR 没有为 Read 的每个部分找到精确匹配的序列,则之前的 MMP 将被扩展。
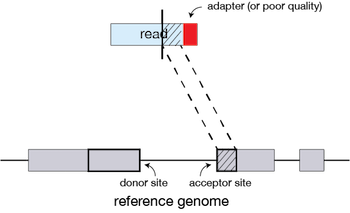
- 如果延伸没有给出良好的比对,那么质量差或接头序列(或其他污染序列)将被软剪切。
聚类、拼接和评分
基于与一组‘anchor’种子或非多重映射种子的接近程度将种子聚集在一起,将单独的种子聚集在一起以创建完整的读取。
然后根据读取的最佳对齐方式将种子拼接在一起(基于不匹配、插入缺失、间隙等进行评分)。
https://github.com/alexdobin/STAR
https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf
https://academic.oup.com/bioinformatics/article/29/1/15/272537
https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/03_alignment.html
 微信
微信 支付宝
支付宝






